Are Metals Made from Molecules?
M.E. Eberhart Structural Chemistry, 28, 1409–1417 (2017)
In the traditional view, covalently bound materials differ in a fundamental way from metallic substances. Though both are built from more basic units that are, in turn, constructed from a small number of atoms, for these two materials classes the nature of these units is thought to be quite different. For covalent solids and liquids, these units are considered to be molecular, meaning that they possess properties and bonding that are retained in the condensed phase and thus they continue to be identifiable within the larger system. For metallic materials, these basic units are considered to be mere constructs that are not observable against the delocalized bonding of metals or alloys. The perceived dissimilarity of metallic and covalently bound materials has fostered distinctly different approaches to their design and improvement. Here, the delocalized view of metallic bonding is examined. This examination suggests that much of the rationale used in the design of molecular materials my be applied to metals and alloys as well.
Quantified Electrostatic Preorganization in Enzymes Using the Geometry of the Electron Charge Density
A. Morgenstern, M. Jaszai, M.E. Eberhart and A.N. Alexandrova Chemical Sciences, 8, 5010-5018, (2017)
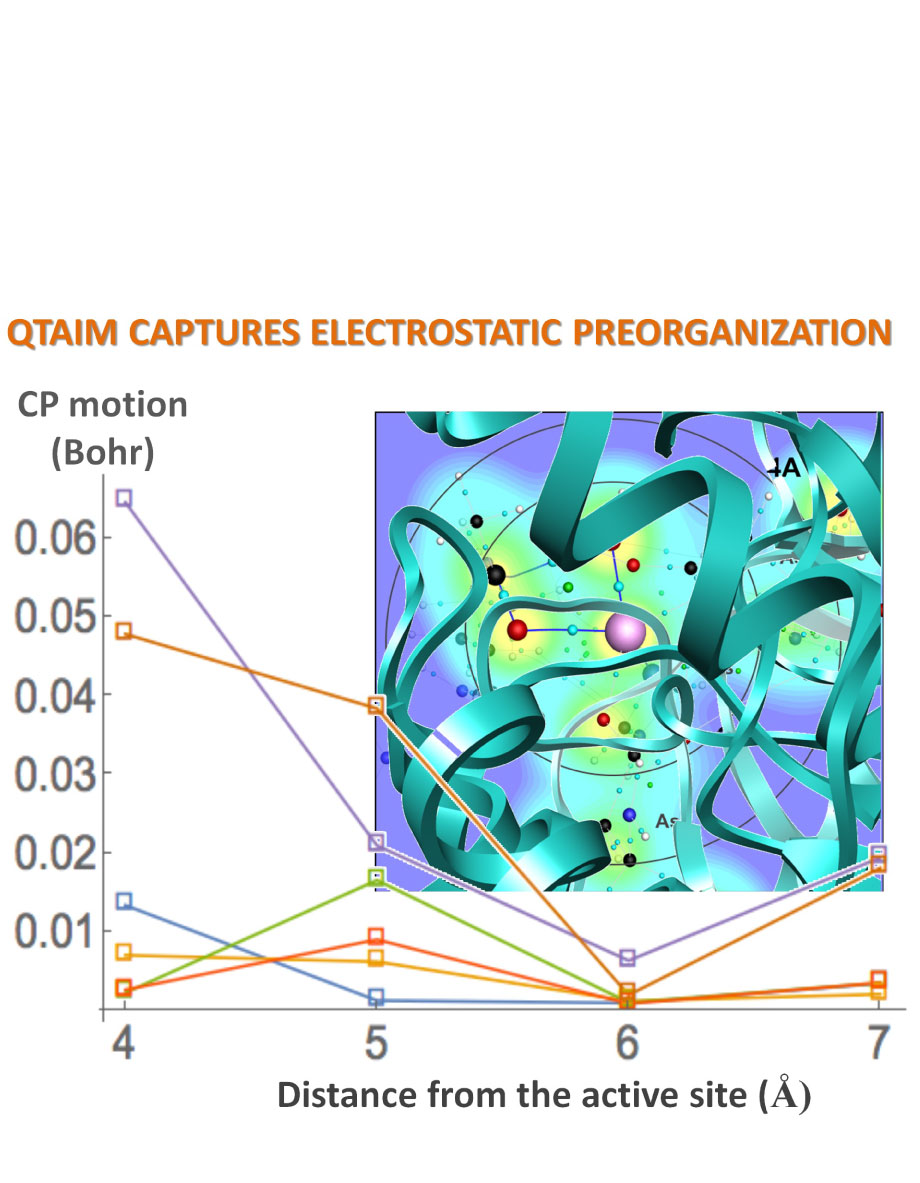
Electrostatic preorganization is thought to be a principle factor responsible for the impressive catalytic capabilities of enzymes. The full protein structure is believed to facilitate catalysis by exerting a highly specific electrostatic field on the active site. Computationally determining the extent of electrostatic preorganization is a challenging process. We propose using the topology and geometry of the electron charge density in the enzyme's active site to asses the effects of electrostatic preorganization. In support of this approach we study the convergence of features of the charge density as the size of the active site model increases in Histone Deacetylase 8. The magnitude of charge density at critical points and most Bader atomic charges are found to converge quickly as more of the protein is included in the simulation. The exact position of critical points however, is found to converge more slowly and be strongly influenced by the protein residues that are further away from the active site. We conjecture that the positions of critical points are affected through perturbations to the wavefunctions in the active site caused by dipole moments from amino acid residues throughout the protein. We further hypothesize that electrostatic preorganization, from the point of view of charge density, can not be easily understood through the charges on atoms or the nature of the bonding interactions, but through the relative positions of critical points that are known to correlate with reactivity and reaction barriers.
Predicting Chemical Reactivity from Charge Density through Gradient Bundle Analysis: Moving beyond Fukui Functiions
A. Morgenstern, T.R. Wilson and M.E, Eberhart Journal of Physical Chemistry A, 121, 4341-4351 (2017)
Predicting chemical reactivity is a major goal of chemistry. Toward this end, atom condensed Fukui functions of conceptual density functional theory have been used to predict which atom is most likely to undergo electrophilic or nucleophilic attack, providing regioselectivity information. We show that the most probable regions for electrophilic attack within each atom can be predicted through analysis of gradient bundle volumes, a property that depends only on the charge density of the neutral molecules. We also introduce gradient bundle condensed Fukui functions to compare the stereoselectivity information obtained from gradient bundle volume analysis. We demonstrate this method using the test set of molecular fluorine, oxygen, nitrogen, carbon monoxide, and hydrogen cyanide.
Nearsightedness of Oxygen-Containing Functional Groups
J. Miorelli and M.E. Eberhart Journal of Physical Chemistry A, 120,9579-9587, (2016)
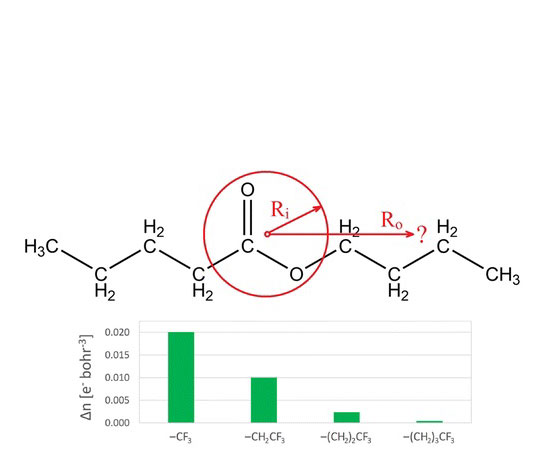
Matter is nearsighted, that is, for a fixed chemical potential, the charge density is only sensitive to perturbations within a radius R. While it is known that the resultant change in the density at point r0 from some
perturbation at some other point R (Δn(r0,R)) is a monotonically decreasing function, a plausible range of a chemically significant Δn(r0,R) and the value of R needed to cause these perturbations has not been well studied. Using the functional group, which upon satisfying the necessary atoms/ bonds specific to that functional group retains a characteristicchemistry, this paper provides an initial study into the magnitude of both Δn and R, the radius beyond which to affect a given property. Values for Δn are shown to be robust
across a variety of DFT functionals and provide a framework for the transfer of the functional group concept to other disciplines, such as metallurgy.
Predictive Methods for Computational Metalloenzyme Redesign - A Test Case with Carboxypeptidase A
B.C. Valdez, A. Morgenstern, M.E. Eberhart, and A.N. Alexandrova, Physical Chemistry Chemical Physics, 18, 31744-31756 (2016)
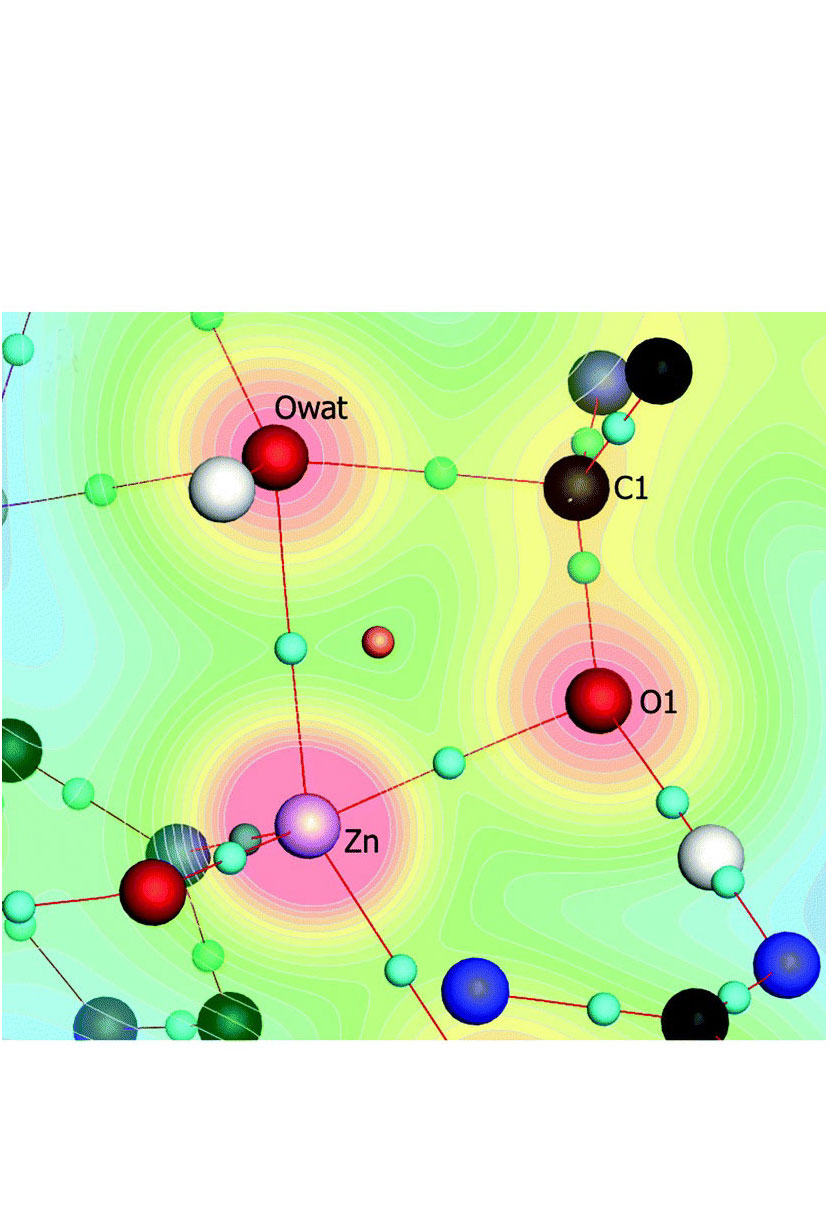
Computational metalloenzyme design is a multi-scale problem. It requires treating the metal coordination quantum mechanically, extensive sampling of the protein backbone, and additionally accounting for the polarization of the active site by both the metal cation and the surrounding protein (a phenomenon called electrostatic preorganization). We bring together a combination of theoretical methods that jointly offer these desired qualities: QM/DMD for mixed quantum-classical dynamic sampling, quantum theory of atoms in molecules (QTAIM) for the assessment of electrostatic preorganization, and Density Functional Theory (DFT) for mechanistic studies. Within this suite of principally different methods, there are both complementarity of capabilities and cross-validation. Using these methods, predictions can be made regarding the relative activities of related enzymes, as we show on the native Zn2+-dependent carboxypeptidase A (CPA), and its mutant proteins, which are hypothesized to hydrolyze modified substrates. For the native CPA, we replicated the catalytic mechanism and the rate in close agreement with the experiment, giving validity to the QM/DMD predicted structure, the DFT mechanism, and the QTAIM assessment of catalytic activity. For most sequences of the modified substrate and tried CPA mutants, substantially worsened activity is predicted. However, for the substrate mutant that contains Asp instead of Phe at the C-terminus, one CPA mutant exhibits a reasonable activity, as predicted across the theoretical methods. CPA is a well-studied system, and here it serves as a testing ground for the offered methods.
The Influence of Zero-Flux Surface Motion on Chemical Reactivity
A. Morgenstern, C. Morgenstern, J. Miorelli, T. Wilson, and M.E. Eberhart, Physical Chemistry Chemical Physics, 91, 5638-5646 (2016)
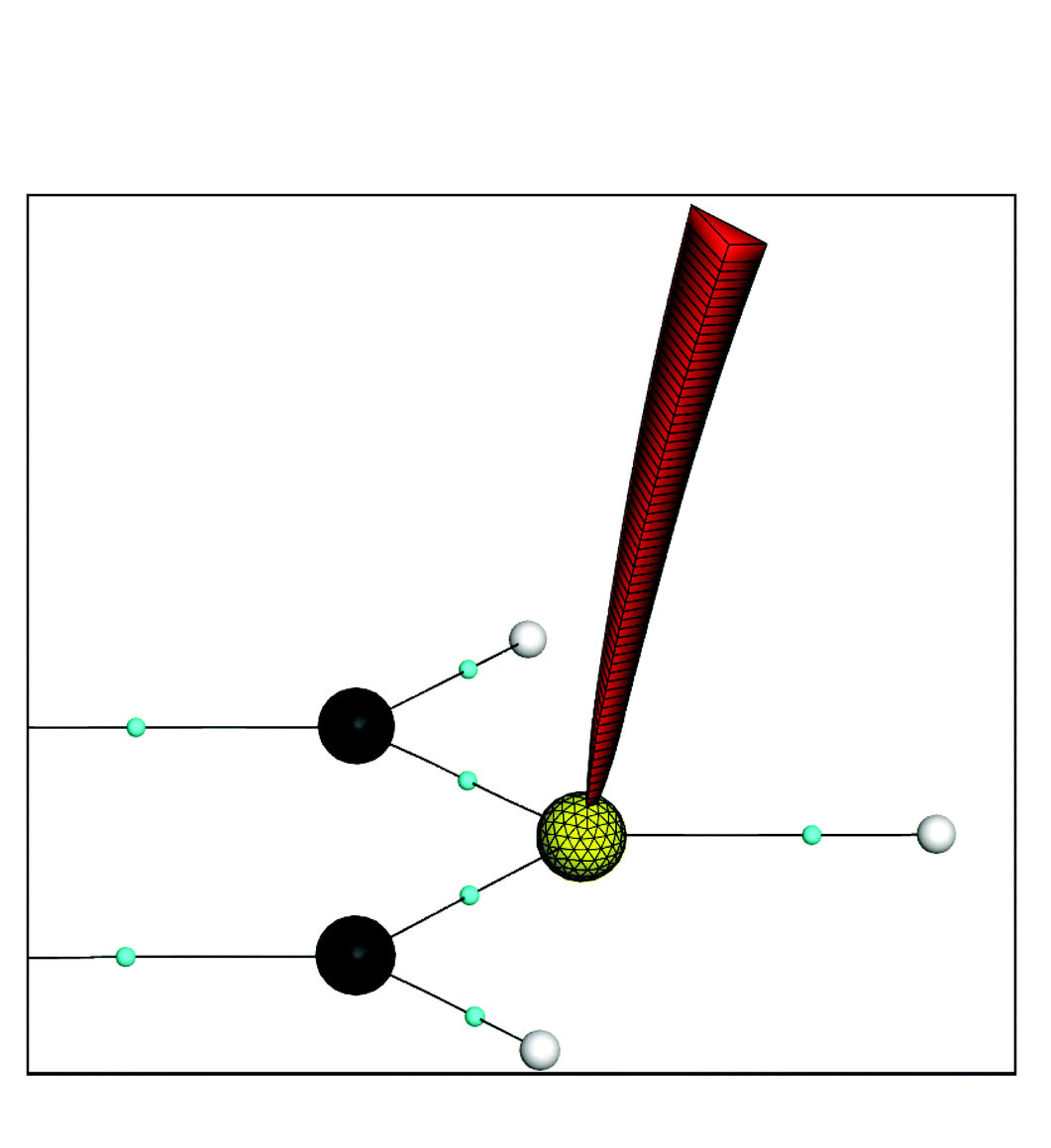
Visualizing and predicting the response of the electron density to an external perturbation provides a portion of the insight necessary to understand chemical reactivity. One strategy used to portray electron response is the electron pushing formalism commonly utilized in organic chemistry, where electrons are pictured as flowing between atoms and bonds. Electron pushing is a powerful tool, but does not give a complete picture of electron response. We propose using the motion of zero-flux surfaces (ZFS) in the gradient of the charge density as an adjunct to electron pushing. Here we derive an equation rooted in conceptual density functional theory showing that the movement of ZFSs contributes to energetic changes in a molecule undergoing a chemical reaction. Using a substituted acetylene, 1-iodo-2-fluoroethyne, as an example, we show the importance of both the boundary motion and the change in electron counts within the atomic basins of the quantum theory of atoms in molecules for chemical reactivity. This method can be extended to study the ZFS motion between smaller gradient bundles in the chrge density in addition to larger atomic basins. Finally, we show that the behavior of the gradient of the charge density within atomic basins contains information about electron response and can be used to predict chemical reactivity.
Bond Dissociation Energies from the Topology of the Charge Density Using Gradient Bundle Analysis
A. Morgenstern and M.E. Eberhart, Physica Scripta, 91, 023012 (2016)
New and more robust models of chemical bonding are necessary to further our understanding of chemical phenomena. Among these are bond bundle and gradient bundle methods, which analyze bonding interactions in terms of property distributions over geometrically defined volumes. These methods have been shown to provide a systematic framework from which to search for structure–property relationships. In addition to providing a brief review of some of the relationships found using this framework, we present new findings that relate the lowering of kinetic energy in bonding regions to bond dissociation energy.
In search of an Intrinsic Chemical Bond
A. Morgenstern, T. Wilson, J. Miorelli, T.E. Jones, and M.E. Eberhart, Computational and Theoretical Chemistry, 1053, 31-37 (2015)
The chemical bond, as a link between atoms, is an intrinsic property of the charge density. However, bond energy, which is commonly seen as the energy difference between a molecular state and an arbitrary dissociated state, depends extrinsically on the charge density. This view of a bond as a natural link possessing properties that are externally determined is at best clumsy, and often leads to contradictory interpretations as to the origins of the structure and properties of molecules and solids. Ideally, one would like to uncover an intrinsic property of the chemical bond that gives similar information content as that provided by bond energy. To this end, we report on our ongoing work exploring the intrinsic geometry imposed on the charge density by mapping it onto the smallest volumes bounded by zero flux surfaces in the gradient of the charge density. These natural volume elements of the Quantum Theory of Atoms in Molecules have well defined properties, and hence, this mapping produces a set of property distributions with a quantifiable geometric structure that varies from molecule to molecule. Here, we examine the intrinsic geometry of the kinetic energy distribution for a series of homonuclear dimers and compare the geometric properties of these distributions with the bond energies of the same dimers.
A Full Topological Analysis of Unstable and Metastable Bond Critical Points
J. Miorelli, T. Wilson, A. Morgenstern, T.E. Jones, and M.E. Eberhart, ChemPhysChem, 16, 152-159 (2015)
Researchers are developing conceptually based models linking the structure and dynamics of molecular charge density to certain properties. Here we report on our efforts to identify features within the charge density that are indicative of instability and metastability. Towards this, we use our extensions to the quantum theory of atoms in molecules that capitalize on a molecule’s ridges to define a natural simplex over the charge density. The resulting simplicial complex can be represented at various levels by its 0-, 1-, and 2-skeleton (dependent sets of points, lines, and surfaces). We show that the geometry of these n-skeletons retains critical information regarding the structure and stability of molecular systems while greatly simplifying charge density analysis. As an example, we use our methods to uncover the fingerprints of instability and metastability in two much-discussed systems, that is, the di-benzene complex and the He and adamantane inclusion complex.
An Electronic Criterions for Assessing Intrinsic Brittleness of Metallic Glasses
X.F. Wang, T.E. Jones, Y. Wu, Z. Lu, S. Halas, T. Durakiewicz, and M.E. Eberhart, Journal of Chemical Physics, 141, 024503 (2014)
Bulk metallic glasses (BMGs) are characterized by a number of remarkable physical and mechan- ical properties. Unfortunately, these same materials are often intrinsically brittle, which limits their utility. Consequently, considerable effort has been expended searching for correlations between the phenomenologically complex mechanical properties of metallic glasses and more basic properties, such correlations might provide insight into the structure and bonding controlling the deformation properties of BMGs. While conducting such a search, we uncovered a weak correlation between a BMG’s work function and its susceptibility to brittle behavior. We argue that the basis for this corre- lation is a consequence of a component of the work function – the surface dipole – and a fundamental bond property related to the shape of the charge density at a bond critical point. Together these ob- servations suggest that simple first principle calculations might be useful in the search for tougher BMGs.
Reactive Cluster Model of Metallic Glasses
T.E. Jones, J. Miorelli, M.E. Eberhart, Journal of Chemical Physics, 140, 84501 (2014)
Though discovered more than a half century ago metallic glasses remain a scientific enigma. Unlike crystalline metals, characterized by short, medium, and long-range order, in metallic glasses short and medium-range order persist, though long-range order is absent. This fact has prompted research to develop structural descriptions of metallic glasses. Among these are cluster-based models that attribute amorphous structure to the existence of clusters that are incommensurate with crystalline periodicity. Not addressed, however, are the chemical factors stabilizing these clusters and promoting their interconnections. We have found that glass formers are characterized by a rich cluster chemistry that above the glass transformation temperature promotes exchange as well as static and vibronic sharing of atoms between clusters. The vibronic mechanism induces correlated motions between neighboring clusters and we hypothesize that the distance over which these motions are correlated mediates metallic glass stability and influences critical cooling rates.
Cauchy Pressure and the Generalized Bonding Model for Nonmagnetic BCC Transition Metals
M.E. Eberhart, and T.E. Jones, Physical Review B, 86, 134106, (2012)
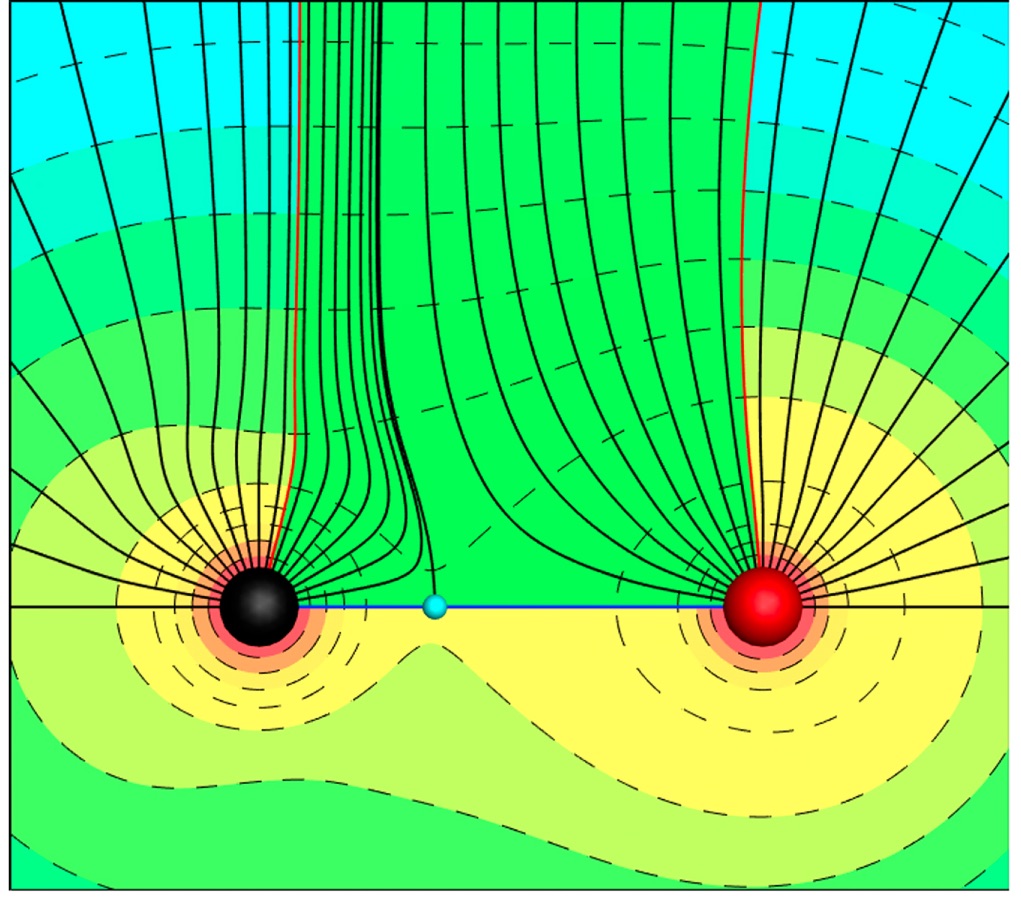
The chemical bond as a physical linkage between atoms has proven to be an immensely powerful heuristic device, providing insight into the general relationships between the structure and properties of molecules and solids. Unfortunately, the bond concept is incomplete and fails to explain more subtle aspects of molecular and materials behavior. A rich set of examples is provided by attempts to identify the structural origins of nonzero Cauchy pressures, a quantity that has been said to reflect the nature of the bonding at the atomic level. We show that these deviations are easily explained by extending the concept of a bond from a linkage between atoms to a set of linkages between charge density critical points (maxima, minima, and saddle points). Further, we show that these links possess an identifiable structure with measurable characteristics. For the nonmagnetic bcc transition metals, we are able to write the Cauchy pressure as a function of these characteristic measures. Significantly, the model should be generalizable to other structures and will provide insight into the relationships between the charge density and elastic response.
Role of Inter- and Intramolecular Bonding on Impact Sensitivity
Travis E. Jones, Journal of Physical Chemistry A, 116, 11008−11014, (2012)
A picture of impact sensitivity based on the bond bundles of the electron charge density is developed, allowing the role of both inter- and intramolecular bonding interactions to be investigated. Impact sensitive materials were found to have a convergent intramolecular bond bundle with a low electron count that serves as a trigger linkage, while insensitive materials do not. The shape and electron count of the intramolecular bond bundles was found to change between the gas phase and solid state due to the formation of intermolecular bond bundles. In the case of polynitrobenzenes, this change was subtle and did not affect the trigger linkages. However, the intermolecular bond bundles in crystalline RDX (hexahydro-1,3,5-trinitro-1,3,5-triazine) change from C−N trigger linkages in the gas phase to N−N trigger linkages in the solid state. This observation offers a theoretical justification of the experimentally observed differences in the decomposition behavior of gas phase and crystalline RDX.
Better Alloys with Quantum Design
T.E. Jones, M.E. Eberhart, S. Imally, C. Mackey, and G.B. Olson, Physical Review Letters, 109, 125506, (2012)
Alloy discovery and development is slowed by trial and error methods used to identify beneficial alloying elements. This fact has led to suggestions that integrating quantum theory and modeling with traditional experimental approaches might accelerate the pace of alloy discovery. We report here on one such effort, using advances in first principles computation along with an evolving theory that allows for the partitioning of charge density into chemically meaningful structures, alloying elements that improve the adhesive properties of interfaces common to high strength steels have been identified.
Bond Bundles and the Origin of Functionality
T.E. Jones, M.E. Eberhart, S. Imally, and C. Mackey, Journal of Physical Chemistry A, 115, 12582, (2011)
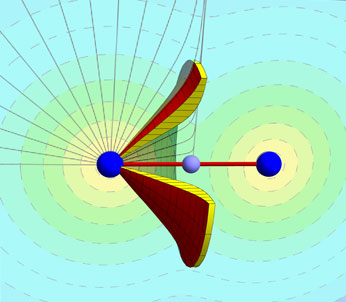
We briefly review the method by which the electron charge density of atomic systems is decomposed into unique volumes called bond bundles, which are characterized by well-defined and additive properties. We then show that boundaries of bond bndles topologically constrain their chemical reactivity. To illustrate this fact, we model the response of the bond bundles of ethane and ethene to electrophilic attack and from the results of these models posit that functional group properties can be inferred from the shapes of their bond bundles. By relating functionality to bond bundle shape, it is possible to see subtle changes in chemical reactivity that are otherwise difficult to explain, as is illustrated by comparing bond bundles through a series of impact sensitive polynitroaromatic molecules.
Topological Catastrophe and Isostructural Phase Transition in Calcium
T.E. Jones, M.E. Eberhart, and D.P. Clougherty, Physical Review Letters, 105, 265702 (2010)
We predict a quantum phase transition in fcc Ca under hydrostatic pressure. Using density functional theory, we find, at pressures below 80 kbar, the topology of the electron charge density is characterized by nearest neighbor atoms connected through bifurcated bond paths and deep minima in the octahedral holes. At pressures above 80 kbar, the atoms bond through non-nuclear maxima that form in the octahedral holes. This topological change in the charge density softens the C0 elastic modulus of fcc Ca, while C44 remains unchanged. We propose an order parameter based on applying Morse theory to the charge density, and we show that near the critical point it follows the expected mean-field scaling law with reduced pressure.
The Bond Bundle in Open Systems
T.E. Jones and M.E. Eberhart, International Journal of Quantum Chemistry, 110, 1500–1505 (2010)
Much of modern chemistry is concerned with the properties and dynamics of chemical bonds. Although they have been described variously, the most familiar representation is that of a link connecting two atoms. However, no one has yet developed a scheme by which to partition a molecule into bond volumes with well-defined properties. As a consequence, the chemical bond is left as nothing more than a heuristic devise. Here, we show molecules can be partitioned into bond-bundles–volumes that share many of the properties associated with the conceptual bond. This partitioning follows naturally through an extension of Baders topological theory of molecular structure. Surprisingly, it also bounds regions of space containing nonbonding or lone-pair electrons and leads to bond orders consistent with those expected from theories of directed valance.
The Irreducible Bundle: Further Structure in the Kinetic Energy Distribution
T.E. Jones, and M.E. Eberhart, Journal of Chemical Physics, 130, 204108 (2009)
Modern molecular sciences are often concerned with the properties and dynamics of chemical bonds. With the ability to experimentally measure charge density, there is a pressing need to find the relationships between charge density and the properties of chemical bonds. Here we show molecules can be partitioned into unique bond volumes characterized by well defined properties. Moreover, this partitioning recovers unrecognized structure in the kinetic energy distribution.
A Quantum Description of the Chemical Bond
M.E. Eberhart, Philososphical Magazine B, 81, 721 (2001)
Traditionally a molecule’s structure and properties are seen as originating
from its bonds, despite the fact that there is no rigorous quantum mechanical description of a bond. Here, the formalism of atoms in molecules is extended and the chemical bond is rigorously defined. Using this definition, it is possible to associate properties unambiguously with particular bonds, with its energy being one such property. With this definition for a bond, it is demonstrated that there are relationships between the bonding and the solid-state properties of a series of transition-metal aluminides. It is shown that these relationships correlate well with the computed difference between first- and second-neighbour bond energies. It is argued that the resulting bonding-property relationships provide the conceptual basis from which to alter bonds predictably and systematically and the properties that originate from these.